Фізико-хімічні методи аналізу (ФХМА) засновані на використанні залежності між вимірюваними фізичними властивостями речовин та їх якісним та кількісним складом. Оскільки фізичні властивості речовин вимірюються з допомогою різних приладів – «інструментів», ці методи аналізу називають також інструментальними методами.
Найбільше практичного застосування серед ФХМА мають:
- електрохімічні методи– ґрунтуються на вимірі потенціалу, сили струму, кількості електрики та інших електричних параметрів;
- спектральні та інші оптичні методи– засновані на явищах поглинання чи випромінювання електромагнітного випромінювання (ЕМІ) атомами чи молекулами речовини;
- хроматографічні методи– ґрунтуються на сорбційних процесах, що протікають у динамічних умовах при спрямованому переміщенні рухомої фази щодо нерухомої.
До переваг ФХМА можна віднести високу чутливість і низьку межу виявлення - масову до 10-9 мкг і концентраційну до 10-12 г/мл, високу селективність (виборчість), що дозволяє визначати компоненти сумішей без їх попереднього поділу, а також експресність проведення аналізів, можливість їх автоматизації та комп'ютеризації.
В аналітичній хімії широко застосовуються електрохімічні методи. Вибір методу аналізу конкретного об'єкта аналізу визначається багатьма чинниками, зокрема, насамперед, нижньою межею визначення елемента.
Дані про нижню межу виявлення різних елементів деякими методами представлені у таблиці.
Межі визначення (мкг/мл) елементів різними методами
| Елемент | МАС | ААС | ПТП | Верба | Іонометрія | Ампером.титрів. |
| Ag | 0,1- дитизон | 0,07 | 0,2 | 0.00001 | 0.02 | 0.05 |
| As | 0,05 - молібд. | 0,2 | 0,04 | 0,02 | - | 0,05 |
| Au | 0,04-метил.фіол. | 0,3 | 0,005 | 0,001 | - | 0,05 |
| Bi | 0,07-дітізон | 0,005 | 0,00001 | - | 0,5 | |
| Cd | 0,04-дітізон | 0,05 | 0,002 | 0,00001 | 0,03 | 0,5 |
| Cr | 0,04-дифе-нілкарбазид | 0,2 | 0,02 | - | - | |
| Cu | 0,03-дітізон | 0,2 | 0,002 | 0,00002 | 0,01 | 0,05 |
| Hg | 0,08-дітізон | - | 0,00005 | |||
| Pb | 0,08-дітізон | 0,6 | 0,003 | 0,00002 | 0,03 | |
| Sb | 0,08-родамін | 0,004 | 0,00004 | - | 0,5 | |
| Fe | 0,1-роданід | 0,2 | 0,003 | 0,0002 | 0,3 | 0,5 |
| Se | 0,08-діамі-нофталін | 0,3 | 0,2 | 0,00002 | - | 0,5 |
| Sn | 0,07-феніл-флуріом | 0,4 | 0,003 | 0,00004 | - | 0,5 |
| Te | 0,1-вісмутол | 0,7 | 0,02 | - | - | |
| Tl | 0,06-родамін | 0,6 | 0,01 | 0,00002 | - | 0,5 |
| Zn | 0,02-дітізон | 0,02 | 0,003 | 0,0003 | - | 0,5 |
| F - | - | - | - | - | 0,02 | 5-10 |
| NH 4 + ,NO 3 - | - | - | - | - | 0,1 | 1-5 |
МАС – молекулярна абсорбційна спекрометрія (фотометрія);
ААС – атомно-абсорбційна спектрометрія (полум'яна фотометрія);
ПТП - змінно-струмова полярографія;
ІВА – інверсійна вольтамперометрія.
Похибки визначень у ФХМА становлять близько 2-5%, проведення аналізів потребує застосування складної та дорогої апаратури.
Розрізняють прямі та непряміметоди фізико-хімічного аналізу У прямих методах використовують залежність величини вимірюваного аналітичного сигналу від концентрації компонента, що визначається. У непрямих методах аналітичний сигнал вимірюють з метою знаходження кінцевої точки титрування визначеного компонента відповідним титрантом, тобто використовують залежність параметра, що вимірюється від об'єматитранта.
Електрохімічні методи аналізузасновані на вивченні та використанні процесів, що протікають на поверхні електрода або в приелектродному просторі. Будь-який електричний параметр (потенціал, електричний струм, кількість електрики та ін), функціонально пов'язаний з концентрацією компонента, що визначається і піддається правильному вимірюванню, може служити аналітичним сигналом.
За природою вимірюваного аналітичного сигналу електрохімічні методи аналізу поділяють на потенціометрію, вольтамперометрію, кулонометріюта ряд інших методів:
Характеристична залежність електрохімічного сигналу від незалежної змінної
| Метод | Вимірюваний сигнал | Залежність сигналу від незалежної змінної |
| Потенціометрія, іонометрія | потенціал E = f(C) С-концентрація аналізованої речовини | |
| Потенціометричне титрування | потенціал E = f(V), V-об'єм реагенту-титранта |
 |
| полярографія, вольтамперометрія | струм I = f(E), E – потенціал поляризації електрода |  |
| інверсійна вольтамперометрія | струм I n = f(E) |  |
| хронопотенціометрія | потенціал E = f (t), t - час поляризації електрода при I = const. |
 |
| амперометричне титрування з одним індикаторним електродом | струм I = f(V), V – об'єм реагенту-титранта |  |
| амперометричне титрування з двома індикаторними електродами | струм I = f(V) V – обсяг реагенту-титранту | |
| кулонометрія | Q = f(C), С – кількість речовини |  |
| кондуктометрія | G = f(C), С – концентрація іонів у розчині |  |
| кондуктометричне титрування | електропровідність G = f(V), V – обсяг реагенту-титранта |  |
Потенціометрія
В основі потенціометричних вимірів лежить залежність рівноважного потенціалу електрода від активності (концентрації) іона, що визначається. Для вимірювань необхідно скласти гальванічний елемент із відповідного індикаторного електрода та електрода порівняння,а також мати прилад для вимірювання потенціалу індикаторного електрода (ЕРС гальванічного елемента), в умовах близьких до термодинамічних, коли індикаторний електрод має рівноважний (або близький до нього) потенціал, тобто без відведення помітного струму від гальванічного елемента при замиканні ланцюга. При цьому не можна використовувати звичайний вольтметр, а слід застосовувати потенціометр- електронний прилад з великим вхідним опором (1011 - 1012 Ом), що унеможливлює перебіг електродних електрохімічних реакцій та виникнення струму в ланцюзі.
Індикаторний електрод - це електрод, потенціал якого залежить від активності (концентрації) іона, що визначається в аналізованому розчині.
Електрод порівняння - це електрод, потенціал якого в умовах проведення аналізу залишається незмінним. По відношенню до електрода порівняння вимірюють потенціал індикаторного електрода Е(ЕРС гальванічного елемента).
У потенціометрії використовують два основні класи індикаторних електродів – електронообмінні та іонообмінні.
Електронообмінні електроди– це електроди, поверхні яких протікають електродні реакції з участю електронів. До таких електродів відносяться електроди першого та другого роду, окислювально-відновлювальні електроди.
Електроди першого роду– це електроди, оборотні по катіону, спільно з матеріалом електрода, наприклад, метал М, занурений у розчин солі того ж металу. На поверхні такого електрода протікає оборотна реакція M n+ + ne↔ M та його реальний потенціал залежить від активності (концентрації) катіонів металу в розчині відповідно до рівняння Нернста:

Для температури 250С (298 K) та для умов, коли активність іонів приблизно дорівнює концентрації (γ → 1):

Електроди першого роду можуть бути виготовлені з різних металів, наприклад, Ag (срібний), Cu (мідний), Zn (цинковий), Pb (свинцевий) та ін.
Схематично електроди першого роду записують як М| M n+ , Де вертикальною лінією показана межа твердої (електрод) і рідкої (розчин) фаз. Наприклад, срібний електрод, занурений у розчин нітрату срібла, зображують наступним чином – Ag | Ag+; за необхідності вказують концентрацію електроліту – Ag | AgNO 3 (0,1 М).
До електродів першого роду відноситься і газовий водневий електрод Pt(H2) | H+ (2Н++ 2е↔ Н 2 , Е 0 = 0):

Електроди другого роду– це електроди, оборотні по аніону, наприклад, метал, покритий малорозчинною сіллю цього металу, занурений у розчин, що містить аніон цієї малорозчинної солі M, MA | А n-. На поверхні такого електрода протікає оборотна реакція MА + ne↔ M + А n-і його реальний потенціал залежить від активності (концентрації) аніону в розчині відповідно до рівняння Нернста (при Т= 298 K і γ → 1):

Прикладами електродів другого роду є хлорсрібний (AgCl + e↔ Ag + Cl -) та каломельний (Hg 2 Cl 2 + 2e↔ 2Hg + 2Cl -) електроди:

Окисно-відновлювальні електроди- Це електроди, які складаються з інертного матеріалу (платина, золото, графіт, скловуглець та ін), зануреного в розчин, що містить окислену (Ок) і відновлену (Вос) форми визначається речовини. На поверхні такого електрода протікає оборотна реакція Ок + ne↔ Вос та його реальний потенціал залежить від активності (концентрації) окисленої та відновленої форм речовини у розчині відповідно до рівняння Нернста (при Т= 298 K і γ → 1):

Якщо електродної реакції беруть участь іони водню, їх активність (концентрацію) враховують у відповідних рівняннях Нернста кожному за конкретного випадку.
Іонообмінні електроди- Це електроди, на поверхні яких протікають іонообмінні реакції. Такі електроди називають також іонселективними чи мембранними.Найважливішою складовою таких електродів є напівпроникна мембрана- тонка тверда або рідка плівка, що відокремлює внутрішню частину електрода (внутрішній розчин) від аналізованого і має здатність пропускати тільки іони одного виду Х (катіони або аніони). Конструктивно мембранний електрод складається з внутрішнього електрода порівняння (зазвичай хлорсрібний) і внутрішнього розчину електроліту з постійною концентрацією потенціального іона, відокремлених від зовнішнього (досліджуваного) розчину чутливою мембраною.
Реальний потенціал іонселективних електродів, виміряний щодо якогось електрода порівняння, залежить від активності тих іонів у розчині, які сорбуються мембраною:

де const –константа, яка залежить від природи мембрани ( потенціал асиметрії) та різниці потенціалів зовнішнього та внутрішнього електродів порівняння, nі а(Х n±) – заряд та активність потенциалопределяющего іона. Якщо потенціал іонселективного електрода виміряно щодо стандартного водневого електрода, то константа є стандартним електродним потенціалом Е 0.
Для мембранних електродів значення крутизни електродної функціїможе відрізнятися від теоретичної нернстівськійвеличини (0,059); у цьому випадку реальне значення електродної функції θ визначають як тангенс кута нахилу градуювального графіка. Тоді:

Потенціал мембранного електрода в розчині, що містить крім визначеного іона Х сторонній іон, що впливає на потенціал електрода, описується рівнянням Микільського(Модифіковане рівняння Нернста):

де z- Заряд стороннього (заважає) іона, KХ/В – коефіцієнт селективності мембранного електрода.
Коефіцієнт селективності KХ/В характеризує чутливість мембрани електрода до визначених іонів Х у присутності іонів, що заважають. Якщо KХ/В<1, то электрод селективен относительно ионов Х и, чем меньше числовое значение коэффициента селективности, тем выше селективность электрода по отношению к определяемым ионам и меньше мешающее действие посторонних ионов. Если коэффициент селективности равен 0,01, то это означает, что мешающий ион В оказывает на величину электродного потенциала в 100 раз меньшее влияние, чем определяемый ион той же молярной концентрации.
Розраховують коефіцієнт селективності як відношення активностей (концентрацій) визначається і заважає іонів, при яких електрод набуває однакового потенціалу в розчинах цих речовин, з урахуванням їх зарядів:

Знаючи значення коефіцієнта селективності можна розрахувати концентрацію іону, що заважає, що впливає на потенціал іонселективного електрода (приклад).
приклад.Яку концентрацію нітратних іонів потрібно створити в 1∙10-3 М розчині фториду натрію, щоб іонселективний фторидний електрод був однаково чутливий до обох іонів, якщо його коефіцієнт селективності електрода?

Рішення.
Так, то
Це означає, що концентрація нітратних іонів в аналізованому розчині понад 0,5 моль/л значно впливає на визначення фторид-іону в його милімо-лярних розчинах.
Класичним прикладом іонселективного електрода з твердою мембраною є скляний електрод з водневою функцією, який служить для вимірювання концентрації іонів водню у розчині (скляний рН-електрод). Для таких електродів мембраною служить спеціальне скло певного складу, а внутрішнім електролітом – 0,1 М розчин хлороводневої кислоти:
Ag, AgCl | 0,1 М HCl | скляна мембрана досліджуваний розчин
На поверхні скляної мембрани відбувається іонообмінний процес:
SiO-Na+ (скло) + Н+ (розчин) → -SiO-H+ (скло) + Na+ (розчин)
внаслідок чого встановлюється динамічна рівновага між іонами водню у склі та розчині Н+ (скло) ↔ Н+ (розчин), що призводить до виникнення потенціалу:
E = const + θ lg a(H+) = const – θ pH
Скляний електрод з підвищеним вмістом у мембрані Al2O3 вимірює активність іонів натрію в розчині (скляний Na-електрод, натрійселективний електрод). В цьому випадку внутрішнім розчином служить 0,1 М розчин натрію хлориду:
Ag, AgCl | 0,1 M NaCl | скляна мембрана досліджуваний розчин
На поверхні скляної мембрани натрійселективного електрода встановлюється рівновага між іонами натрію в склі та розчині Na+ (скло) ↔ Na+ (розчин), що призводить до виникнення потенціалу:
E = const + θ lg a(Na+) = const – θ pNa
Найбільш досконалим електродом з кристалічною мембраною є фторидселективний електрод, мембрана якого виконана з пластинки монокристала фториду лантану (LaF3), активованого збільшення провідності фторидом європію (EuF 2):
Ag, AgCl | 0,1 M NaCl, 0,1 M NaF | LaF 3 (EuF 2) | досліджуваний розчин
Потенціал фторидного електрода визначається іонообмінним процесом на його поверхні F-(мембрана) ↔ F-(розчин):
E = const - θ lg a(F-) = const + θ pF
Значення константи та крутості електродної функції θ для іонселективних електродів визначають з градуювального графіка Е÷рХ як відрізок на осі ординат та тангенс кута нахилу прямої відповідно. Для скляного рН-електроду ця операція замінюється налаштуванням приладів (рН-метрів) за стандартними буферними розчинами з точно відомими значеннями рН.
Схематичний вигляд скляного та фторидселективного електродів наведено на рисунках:
У парі з індикаторним електродом для вимірювання його потенціалу (ЕРС гальванічної комірки) використовують електрод порівняння з відомим та стійким потенціалом, що не залежить від складу досліджуваного розчину. Найчастіше як електрод порівняння застосовують хлорсрібний і каломельний електроди. Обидва електроди відносяться до електродів другого роду та характеризуються високою стабільністю в роботі.
Потенціали хлорсрібного та каломельного електродів залежать від активності (концентрації) хлорид-іонів (при Т= 298 K і γ → 1):

Як електроди порівняння найчастіше застосовують електроди з насиченим розчином хлориду калію – при 250С потенціал насиченого хлорсрібного електрода порівняння дорівнює +0,201, а насиченого каломельного +0,247 (відносно стандартного водневого електрода). Потенціали для хлорсрібних та каломельних електродів порівняння, що містять 1 М та 0,1 М розчини хлориду калію, можна знайти у довідкових таблицях.
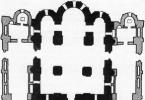
Схематичний вид насичених хлорсрібного та каломельного електродів порівняння наведено на малюнку:

Електроди порівняння хлорсрібний (а)та каломельний (б)
1 - азбестове волокно, що забезпечує контакт з аналізованим розчином
2 - розчин KCl (насичений)
3 - отвір для контакту
4 - розчин KCl (насичений), AgCl (тв.)
5 - отвір для введення розчину KCl
6 - паста із суміші Hg2Cl2, Hg та КС1 (насичений)
Потенціометричний аналіз широко застосовують для безпосереднього визначення активності (концентрації) іонів у розчині шляхом вимірювання рівноважного потенціалу індикаторного електрода (ЕРС гальванічного елемента) – пряма потенціометрія (іонометрія), а також для індикації кінцевої точки титрування ( ктт) щодо зміни потенціалу індикаторного електрода в процесі титрування ( потенціометричне титрування).
У всіх прийомах прямий потенціометріївикористовується залежність індикаторного електрода від активності (концентрації) іона, що визначається, яка описується рівнянням Нернста. Результати аналізу мають на увазі визначення концентрації речовини, а не її активності, що можливо при значенні коефіцієнтів активності іонів рівних одиниці (γ → 1) або їх постійному значенні (постійній іонній силі розчину), тому в подальших міркуваннях використовуються лише концентраційні залежності.
Концентрація іона може бути розрахована за експериментально знайденим потенціалом індикаторного електрода, якщо для електрода відомі постійна складова (стандартний потенціал Е 0) і крутість електродної функції θ . У цьому випадку складається гальванічний елемент, що складається з індикаторного електрода та електрода порівняння, вимірюється його ЕРС, розраховується потенціал індикаторного електрода (щодо СВЕ) та концентрація іона, що визначається.
У методу градуювального графікаготують серію стандартних розчинів з відомою концентрацією іона і постійною іонною силою, вимірюють потенціал індикаторного електрода щодо електрода порівняння (ЕРС гальванічного елемента) в цих розчинах і за отриманими даними будують залежність Е÷ р З(А) (градуювальний графік). Потім вимірюють потенціал індикаторного електрода в аналізованому розчині Ех (у тих самих умовах) і за графіком визначають р Зх(А) і розраховують концентрацію обумовленої речовини в аналізованому розчині.
У метод стандарту (порівняння)вимірюють потенціал індикаторного електрода в аналізованому розчині ( Ех) та у стандартному розчині визначається речовини ( Ест). Розрахунок концентрації іона проводять виходячи з рівнянь Нернста для аналізованої проби і стандартного зразка. Крутизна електродної функції для індикаторного електрода θ
При використанні методу добавокспочатку вимірюють потенціал індикаторного електрода в аналізованому розчині ( Ех), потім додають до нього певний обсяг стандартного розчину визначається речовини і вимірюють потенціал електрода в отриманому розчині з добавкою ( Ех+д). Розрахунок концентрації іона проводять виходячи з рівнянь Нернста для аналізованої проби і проби з добавкою. Крутизна електродної функції для індикаторного електрода θ повинна бути відома або визначена заздалегідь за градуювальним графіком.
При потенціометричному титруваннівимірюють та записують ЕРС електрохімічного осередку (потенціал індикаторного електрода) після додавання кожної порції титранта. Потім за отриманими результатами будують криві титрування – інтегральнуу координатах E ÷ V(а)і диференціальнуу координатах ∆ E/∆V ÷ V (б), і визначають кінцеву точку титрування ( ктт)графічним способом:

У потенціометричному титруванні використовують усі основні типи хімічних реакцій – кислотно-основні, окислювально-відновні, осадження та комплексоутворення. До них пред'являються ті ж вимоги, що і у візуальній титриметрії, доповнені наявністю відповідного індикаторного електрода для фіксації зміни концентрації іонів потенційновизначальних в ході титрування.
Похибка визначення при проведенні потенціометричного титрування становить 0,5-1%, що суттєво нижче, ніж при прямих потенціометричних вимірах (2-10%), проте при цьому спостерігаються вищі межі виявлення – більше 10 -4 моль/л.
Кулонометрія
Кулонометріяпоєднує методи аналізу, що ґрунтуються на вимірі кількості електрики, витраченої на електрохімічну реакцію. Електрохімічна реакція призводить до кількісного електроперетворення (окислення або відновлення) визначуваної речовини на робочому електроді (пряма кулонометрія) або отримання проміжного реагенту (титранта), який стехіометрично реагує з визначальною речовиною (непряма кулонометрія, кулонометричне титрування).
В основі кулонометричних методів лежить закон Фарадея, який встановлює зв'язок між кількістю електроперетвореної (окисленої або відновленої) речовини та кількістю витраченої при цьому електрики:

де m- Маса електроперетвореної речовини, г; Q– кількість електрики, витраченої на електроперетворення речовини, Кл; F– число Фарадея, що дорівнює кількості електрики, необхідної для електроперетворення одного моль-еквівалента речовини, 96500 Кл/моль; М– молярна маса речовини, г/моль; n– число електронів, що у електрохімічної реакції.
Необхідною умовою проведення кулонометричного аналізу є практично повне витрачання електрики на перетворення визначеної речовини, тобто електрохімічна реакція повинна протікати без побічних процесів зі 100% виходом струму.
На практиці кулонометричний аналіз реалізується у двох варіантах – за постійного потенціалу ( потенціостатична кулонометрія) і при постійній силі струму( амперостатичнакулонометрія).
Потенціостатичну кулонометріюзастосовують для прямихкулонометричних вимірювань, коли електроліз піддається безпосередньо визначається речовина. При цьому потенціал робочого електрода за допомогою потенціостатівпідтримується постійним і його значення вибирають на основі кривих поляризаційних в області граничного струму визначається речовини. У процесі електролізу при постійному потенціалі сила струму зменшується відповідно до зменшення концентрації електроактивної речовини за експоненційним законом:

де Ι - Сила струму в момент часу t, А; Ι 0 - сила струму в початковий момент електролізу, А; k- Константа, яка залежить від умов електролізу.
Електроліз ведуть до досягнення залишкового струму Ι , величина якого визначається необхідною точністю – для допустимої похибки 0,1% електроліз вважатимуться завершеним при Ι = 0,001Ι 0 . Для скорочення часу електролізу слід застосовувати робочий електрод великої поверхні при інтенсивному перемішуванні аналізованого розчину.
Загальна кількість електрики Q, необхідне електроперетворення визначається речовини, визначається рівнянням:

Визначити кількість електрики можна виміром площі під кривою «струм – час» за допомогою механічних або електронних інтеграторів або за допомогою хімічних кулонометрів. Кулонометр– це електролітичний осередок, у якому зі 100% виходом по струму протікає електрохімічна реакція відомої стехіометрії. Кулонометр включають послідовно з досліджуваним кулонометричним осередком, тому за час електролізу через обидві осередки протікає однакова кількість електрики. Якщо після закінчення електролізу виміряти кількість (масу) речовини, що утворилася в кулонометрі, то за законом Фарадея можна розрахувати кількість електрики. Найчастіше застосовують срібний, мідний та газові кулонометри.
Амперостатичну кулонометріюзастосовують для кулонометричного титруванняпри постійному струмі, в процесі якого визначається речовина реагує з титрантом, що утворюється в результаті електрохімічної реакції на робочому електроді, а тому званий електрогенерованим титрантом.
Для забезпечення 100%-ного виходу струму необхідний значний надлишок допоміжної речовини, з якого генерується титрант, що виключає перебіг конкуруючих реакцій на робочому електроді. При цьому титрант генерується в кількості, еквівалентній визначеній речовині, і за кількістю електрики, витраченої на генерацію титранта, можна розрахувати вміст речовини, що визначається.
Кількість електрики Qв кулонометрії при постійній силі струму Ι розраховують за формулою:

де t– час електролізу, визначення якого придатні практично всі способи встановлення кінцевої точки в титриметрії (візуальні – індикатори, інструментальні – потенціометрія, амперометрія, фотометрія). При силі струму в амперах та часі електролізу в секундах отримуємо кількість електрики в кулонах (приклад).
приклад.На кулонометричне титрування розчину аскорбінової кислоти йодом, що генерується з іодиду калію струмом силою 5,00 мА, знадобилося 8 хв 40 с. Розрахувати масу аскорбінової кислоти в аналізованому розчині. Запропонувати спосіб фіксування кінцевої точки титрування.
Рішення.Кількість електрики, витрачена на окислення йодиду і, відповідно, аскорбінової кислоти дорівнює:
Q = Ι·t= 5,00 · 10 -3 · 520 = 2,60 Кл.
Аскорбінова кислота окислюється йодом до дегідроаскорбінової кислоти з віддачею двох електронів (С6Н8О6-2 е→ З 6 Н 6 Про 6 + 2Н +), тоді за законом Фарадея:

Кінцева точка титрування визначається за появою надлишку йоду в розчині. Отже, фіксувати її можна візуально за допомогою крохмалю, доданого в аналізований розчин (поява синього забарвлення), амперометрично з ртутним крапельним або платиновим мікроелектродом по появі граничного струму йоду, потенціометрично по різкому збільшенню потенціалу платинового електрода.
Вольтамперометрія
Вольтамперометричний метод аналізузаснований на використанні явища поляризації мікроелектроду, одержанні та інтерпретації вольтамперних (поляризаційних) кривих, що відображають залежність сили струму від прикладеної напруги. Вольтамперна крива (вольтамперограма) дозволяє одночасно отримати якісну та кількісну інформацію про речовини, що відновлюються або окислюються на мікроелектроді (деполяризаторах), а також про характер електродного процесу. Сучасна вольтамперометрія – високочутливий та експресний метод визначення речовин, придатний для аналізу різних об'єктів неорганічної та органічної природи, у тому числі й фармацевтичних препаратів. Мінімально визначається концентрація у вольтамперометрії досягає значень 10 -8 моль/л при похибці методу менше 5%. Вольтамперометрія за оптимальних умов експерименту дозволяє в аналізованому розчині визначати кілька компонентів одночасно.
У вольтамперометрії використовують два електроди – робітникполяризований електрод з малою поверхнею (індикаторний мікроелектрод) і допоміжнийнеполяризується електрод з великою поверхнею (електрод порівняння). Робочими електродами служать мікроелектроди з ртуті (ртутний капаючий електрод, РКЕ), платини (ПЕ) та вуглецевих струмопровідних матеріалів (графіт, скловуглець).
При проходженні постійного струму через електролітичну комірку процес характеризується співвідношенням (закон Ома для розчину електроліту):
Е = Ea - Eк + IR
Де Е– додана зовнішня напруга; Еа- Потенціал анода; Ек- Потенціал катода; I- Струм у ланцюгу; R- Внутрішній опір електролітичного осередку.
При вольтамперометричних вимірах аналізований розчин містить індиферентний (фоновий) електроліт великої концентрації (в 100 разів і більше перевищує концентрацію визначається речовини – опір розчину мало), а струм у вольтамперометрії не перевищує 10 -5 А, тому падінням напруги в комірці IRможна знехтувати.
Оскільки в вольтамперометрії один з електродів (допоміжний) не поляризується і для нього потенціал залишається постійним (його можна прийняти рівним нулю), напруга, що подається на комірку, проявляється в зміні потенціалу тільки робочого електрода і тоді Е = Eaдля робочого мікроаноду ( анодна поляризація) та Е =-Екдля робочого мікрокатода ( катодна поляризація). Таким чином, вольтамперна крива, що реєструється, відображає електрохімічний процес, що відбувається тільки на робочому електроді. Якщо в розчині присутні речовини, здатні електрохімічно відновлюватися або окисляться, то при накладенні на комірку напруги, що лінійно змінюється, вольтамперограма має форму хвилі 1 (у відсутності електрохімічної реакції залежність струму від напруги лінійна 2 відповідно до закону Ома):

Розділ вольтамперометрії, в якому робочим мікроелектродом служить РКЕ називають полярографією, на честь чеського електрохіміка Я.Гейровського, який запропонував цей метод у 1922 році. Вольтамперограми, отримані в осередку з ртутним електродом, що капає, називають полярограмами.
Для реєстрації класичних полярограм осередок з РКЕ (робочий електрод) та насиченим каломельним електродом (допоміжний електрод, електрод порівняння) приєднують до джерела постійної напруги та змінюють потенціал зі швидкістю 2-5 мВ/с.
Ртутний капаючий електрод є практично ідеально поляризується в широкому діапазоні потенціалів, обмеженому в анодній області електродними реакціями окислення ртуті (+0,4 В), а в катодної реакціями відновлення іонів водню (від -1 до -1,5 В залежно від кислотності середовища) або катіонів фону (від -2 для катіонів лужних металів до -2,5 для R 4 N +). Це дозволяє вивчати та визначати на РКЕ речовини, що відновлюються при дуже високих негативних потенціалах, що неможливо на електродах з інших матеріалів. Слід зазначити, що тут і далі значення потенціалів наведені щодо насиченого каломельного електрода і при необхідності можуть бути перераховані по відношенню до іншого порівняного електроду, наприклад, насиченого хлорсрібного.
Перед реєстрацією полярограми на РКЕ необхідно видалити розчинений кисень, оскільки він є електроактивним в негативній області потенціалів, даючи дві хвилі відновлення при -0,2 і -0,9 В. Зробити це можна, насичуючи розчин інертним газом (азот, аргон, гелій). З лужних розчинів кисень видаляють за допомогою сульфіту натрію (O 2 + 2Na 2 SO 3 → 2Na 2 SO 4).
Класична полярограма (полярографічна хвиля) в ідеалізованому вигляді представлена нижче:

Основними характеристиками полярографічної хвилі є величина дифузійного струму ( Iд, мкА), потенціал напівхвилі ( Е 1/2 , В) – потенціал, при якому струм дорівнює половині дифузійної, і нахил висхідної ділянки (0,059/ n– крутість електродної функції). Ці параметри дозволяють використовувати полярографію як метод аналізу (сила струму пропорційна концентрації) та дослідження (потенціал напівхвилі та електродна функція залежать від природи речовини).
На початковій ділянці полярографічної хвилі (А-Б) струм із зміною потенціалу зростає дуже повільно – це так званий залишковий струм (Iост) . Основний внесок у залишковий струм робить формування подвійного електричного шару ( струм заряджання), який неможливо виключити та величина якого зростає зі збільшенням потенціалу. Другим складовим залишкового струму є струм, обумовлений електроактивними домішками, який можна зменшити застосовуючи чисті реактиви та воду.
При досягненні точки Б ( потенціал виділення– при відновленні на катоді потенціал виділення називають потенціалом відновлення Евісь, при окисленні на аноді – потенціалом окислення Еок) на електроді починається електрохімічна реакція, в яку вступає електроактивна речовина (деполяризатор), у результаті струм різко зростає (ділянка Б-В) до деякого граничного значення, залишаючись потім практично постійним (ділянка В-Г). Струм, що відповідає цій ділянці називають граничним струмом(Iпр), а різниця між граничним та залишковим струмом становить дифузійний струм (Iд = Iпр – Iзуп). На ділянці В-Г зі збільшенням потенціалу граничний і залишковий струми незначно зростають, а значення дифузійного струму залишається незмінним. Підйом струму у точці Р обумовлений новою електрохімічною реакцією (наприклад, відновленням катіонів фонового електроліту).
Дифузійний струм отримав свою назву внаслідок того, що в цій галузі потенціалів в результаті електрохімічної реакції в приэлектродном шарі спостерігається практично повна відсутність деполяризатора та його збагачення речовиною відбувається за рахунок дифузії деполяризатора з глибини розчину, де його концентрація залишається постійною. Оскільки швидкість дифузії в даних конкретних умовах залишається постійною, то дифузійний струм зберігає сталість свого значення.
Залежність величини дифузного струму від концентрації деполяризатора для р.к.е. виражається рівнянням Ільковича:
I d = 605nD 1/2 m 2/3 t 1/6 c
де D – коефіцієнт дифузії електроактивного іона; n – число електронів, що у реакції; m 2/3 t 1/6 – характеристика капіляра, з якого випливає ртуть; с - концентрація речовини, що визначається (деполяризатора).
При роботі з одним і тим же капіляром та деполяризатором значення 605nD 1/2 m 2/3 t 1/6 = const, тому між висотою хвилі та концентрацією речовини є лінійна залежність
На цій лінійній залежності ґрунтується кількісний полярографічний аналіз. Взаємозв'язок між потенціалом електрода і струмом, що виникає, описується рівнянням полярографічної хвилі (рівняння Ільковича-Гейровського):

де Е та I – відповідно потенціал та величина струму для даної точки полярографічної кривої; I d – величина дифузійного струму; Е 1/2 – потенціал напівхвилі.
Е 1/2 - це потенціал, при якому досягається величина струму, що дорівнює половині I d . Він залежить від концентрації деполяризатора. Е 1/2 дуже близькі до нормального редокс-потенціалу системи (Ео), тобто є якісною характеристикою, що визначається тільки природою іонів, що відновлюються і за якими можна встановити якісний склад аналізованого розчину.
Полярограма (вольтамперограма) містить цінну аналітичну інформацію – потенціал напівхвилі Е 1/2 є якісною характеристикою деполяризатора (якісний аналітичний сигнал), тоді як дифузійний струм Iд лінійно пов'язаний з концентрацією речовини, що визначається, в об'ємі аналізованого розчину (кількісний аналітичний сигнал) – Iд = KС.
Величина Е 1/2 може бути розрахована з рівняння полярографічної хвилі або визначена графічно:

Знайдене значення Е 1/2 з урахуванням використаного фонового електроліту дозволяє виходячи з табличних даних ідентифікувати деполяризатор. Якщо в аналізованому розчині знаходиться кілька речовин, потенціали напівхвиль яких різняться більш ніж на 0,2, то на полярограмі буде не одна хвиля, а кілька - за кількістю електроактивних частинок. При цьому слід мати на увазі, що відновлення (окислення) багатозарядних частинок може відбуватися східчасто, даючи кілька хвиль.
Для виключення переміщення речовини до електрода за рахунок теплової та механічної конвекції (перемішування) вимірювання здійснюється у термостатованому розчині та у відсутності перемішування. Усунення електростатичного тяжіння деполяризатора полем електрода (міграції) сприяє великий надлишок електронеактивного фонового електроліту, іони якого екранують заряд електрода, зменшуючи рушійну силу міграції практично до нуля.
При використанні ртутного електрода, що капає, на полярограмі спостерігаються осциляції струму(його періодичне невелике збільшення та зменшення). Кожна така осциляція відповідає виникненню, зростанню та відриву краплі ртуті від капіляра мікроелектроду. У полярографах передбачено пристрої для усунення осциляцій.
Полярограми можуть бути спотворені за рахунок полярографічних максимумів- різкого зростання струму вище його граничного значення з наступним спадом:

Поява максимумів обумовлена перемішуванням розчину в результаті руху поверхні краплі ртуті через нерівномірний розподіл заряду, а, відповідно, і поверхневого натягу (максимуми I роду), а також появи завихрень при витіканні ртуті з капіляра (максимуми II роду). Максимуми спотворюють полярограму і ускладнюють її розшифрування. Для видалення максимумів I роду вводять поверхнево-активну речовину (наприклад, агар-агар, желатин, камфару, фуксин, синтетичні ПАР), яка, адсорбуючись на поверхні ртутної краплі, вирівнює поверхневий натяг і усуває рух поверхневих шарів ртуті. Для видалення максимумів ІІ роду достатньо зменшити тиск ртуті в капілярі, зменшивши висоту ртутного стовпа.
Вольтамперометрія з твердими робочими електродамивідрізняється від полярографії з використанням РКЕ іншим діапазоном поляризації мікроелектроду. Як було показано вище, ртутний капаючий електрод внаслідок високого перенапруги водню на ньому можна використовувати в області високих негативних потенціалів, але через анодне розчинення ртуті при +0,4 В він не може бути застосований для досліджень в області позитивних потенціалів. На графіті і платині розряд іонів водню протікає значно легше, тому область їхньої поляризації обмежена значно нижчими негативними потенціалами (-0,4 і -0,1 відповідно). У той же час в області анодних потенціалів платиновий та графітовий електроди придатні до потенціалу +1,4 В (далі починається електрохімічна реакція окиснення кисню води 2Н 2 О – 4 е→ Про 2+4Н+), що робить їх придатними для досліджень у діапазоні позитивних потенціалів.
На відміну від РКЕ під час реєстрації вольтамперограми, поверхня твердого мікроелектроду не відновлюється і легко забруднюється продуктами електродної реакції, що призводить до зниження відтворюваності та точності результатів, тому перед реєстрацією кожної вольтамперограми слід проводити очищення поверхні мікроелектроду.
Стаціонарні тверді електроди не знайшли широкого застосування у вольтамперометрії через повільне встановлення граничного струму, що призводить до спотворення форми вольтамперограми, однак, на мікроелектродах, що обертаютьсяу приелектродному шарі виникають умови для стаціонарної дифузії, тому сила струму встановлюється швидко і вольтамперограма має ту саму форму, що й у разі РКЕ.
Величина граничного дифузійного струму на дисковому електроді, що обертається (незалежно від матеріалу) описується рівнянням конвективної дифузії (Левича):
I d = 0.62nFSD 2/3 w 1/2 n -1/6 c
де n - число електронів, що у електродному процесі;
F – число Фарадея (96 500 кулонів);
S – площа електрода;
D – коефіцієнт дифузії деполяризатора;
w - кутова швидкість обертання електрода;
n - кінематична в'язкість досліджуваного розчину;
с – концентрація деполяризатора, моль/л.
При труднощі в розшифровці полярограм застосовують метод «свідка» – після реєстрації полярограми аналізованого розчину, до нього електролітичну комірку по черзі додають стандартні розчини передбачуваних сполук. Якщо припущення було вірним, збільшується висота хвилі відповідної речовини, при неправильному припущенні з'явиться додаткова хвиля при іншому потенціалі.
Визначити концентрацію деполяризатора в аналізованому розчині можна методами градуювального графіка, методом стандарту (порівняння) та методом добавок. При цьому завжди слід використовувати стандартні розчини, склад яких максимально наближений до складу аналізованого розчину, а умови реєстрації полярограм повинні бути однакові. Методи застосовуються в інтервалі концентрацій, де суворо дотримується прямо пропорційна залежність дифузійного струму від концентрації деполяризатора. Насправді при кількісних визначеннях, зазвичай, не фіксують величину дифузійного струму в мкА, а вимірюють висоту полярографічної хвилі h, як зазначено на попередньому малюнку, яка також є лінійною функцією від концентрації h = KC.
за методу градуювального графікареєструють полярограми серії стандартних розчинів та будують градуювальний графік у координатах h ÷ C(або Iд÷ З), за яким для знайденого значення h x в аналізованому розчині знаходять концентрацію обумовленої речовини в ньому Зх.
У метод стандарту (порівняння)в одних і тих же умовах записують полярограми аналізованого та стандартного розчинів визначуваної речовини з концентраціями Зх і Зст, тоді:

При використанні методу добавокспочатку записують полярограму аналізованого розчину об'ємом V x з концентрацією Зх і вимірюють висоту хвилі h x. Потім в електролітичну комірку до аналізованого розчину додають певний об'єм стандартного розчину речовини, що визначається. Vд з концентрацією Зд (переважно, щоб V x>> Vд і Зх<Зд), записують полярограму розчину з концентрацією Зх+д і вимірюють висоту отриманої хвилі hх+д. Нескладні перетворення дозволяють за цими даними дозволяють розрахувати концентрацію речовини, що визначається, в аналізованому розчині (приклад).
приклад.При полярографуванні 10,0 мл нікотинаміду розчину отримана хвиля висотою 38 мм. Після додавання до цього розчину 1,50 мл стандартного розчину, що містить 2,00 мг/мл нікотинаміду, хвиля збільшилася до 80,5 мм. Розрахувати вміст препарату (мг/мл) у аналізованому розчині.
Рішення.Висота хвилі нікотинаміду в аналізованому розчині h x відповідно до рівняння Ільковича дорівнює:

а після добавки стандартного розчину ( hх+д):

Якщо перше рівняння почленно поділити на друге, то отримаємо:

Вирішуючи рівняння щодо Зх і підставивши значення величин із умови завдання.
Опис роботи
Сучасні галузі виробництва та соціального життя людей ставлять свої специфічні завдання перед фізико-хімічними методами аналізу контролю якості продукції. Одним із основних фізико-хімічних методів аналізу є електрохімічні методи аналізу.
Цими методами можна швидко і досить точно визначити багато показників якості продукції.
Електрохімічні методи аналізу складу речовини широко використовують у різних галузях промисловості. Вони дозволяють автоматизувати отримання результатів якості продукції і виправляти порушення, не зупиняючи виробництво. У харчовій промисловості цими методами визначають кислотно-лужний баланс продукту, наявність шкідливих та токсичних речовин та інші показники, що впливають не лише на якість, а й на безпеку їжі.
Обладнання, призначене для проведення електрохімічних аналізів, відрізняється відносною дешевизною, доступністю та простотою у використанні. Тому ці методи мають широке застосування у спеціалізованих лабораторіях, а й у багатьох виробництвах.
У зв'язку з цим метою даної ку
ВСТУП 2
ТЕОРЕТИЧНА ЧАСТИНА 3
1.1 Загальна характеристика фізико-хімічних методів аналізу
1.2 Характеристика електрохімічних методів 4
1.3 Класифікація електрохімічних методів аналізу
2 ЕКСПЕРЕМЕНТАЛЬНО-ПРАКТИЧНА ЧАСТИНА 15
ВИСНОВОК 21
СПИСОК ЛІТЕРАТУРИ 22
У електрохімічних методах вимірювання концентрації використовують електрохімічний осередок. Найпростіша комірка складається з пари електродів, занурених у розчин електроліту. Розчин електроліту поміщений в одну посудину або в дві, з'єднаних між собою містком з електролітом (комірка з перенесенням). Електроди можуть бути з'єднані між собою провідником (внутрішній електроліз) або провідниками через джерело живлення (зовнішній електроліз).
Механізм перенесення електрики у різних ділянках електричного ланцюга різний. По провідникам електричний заряд переноситься електронами, розчині – іонами. На межі розділу фаз відбувається зміна механізму провідності в результаті протікання гетерогенної окисно-відновної реакції. Її називають електрохімічною чи електродною реакцією, тобто реакцією, що з обміном зарядами між хімічними сполуками, що у різних фазах – твердої (поверхня електрода) і рідкої (розчин електроліту).
Є хімічні сполуки в розчині, які легко віддають електрони електроду, виготовленому з певного матеріалу, наприклад, платини або графіту, тобто окислюються на ньому. Такий електрод називають анодом. На поверхні анода утворюється окислювач, який може залишитися (адсорбуватися), розчинитися в матеріалі анода (ртутному аноді) або дифундувати в розчин електроліту під дією сил дифузії (градієнта концентрації).
Наприклад, у розчині CuCl 2
2Cl - - 2 e= Cl 2
(Red 1 - ne= Ox 1)
Розчин Pt-електрод
Cl - → ←Cl 2
Газоподібний Cl 2 , що утворюється на поверхні платинового електрода, дифундуватиме в розчин електроліту.
Є також розчині хімічні сполуки, які легко приймають електрони від електрода, тобто. відновлюються на ньому. Такий електрод називають катодом. На поверхні катода утворюється відновник, який може залишитися (адсорбуватися), розчинитися в матеріалі анода (ртутному катоді) або дифундувати в розчин електроліту під дією сил дифузії.
Наприклад, у розчині CuCl 2
Cu 2+- + 2e = Cu 0
(Ox 2 + ne = Red 2)
розчин Hg-електрод
Cu 2+ → Cu 0 → Cu 0 (Hg)
Атоми міді, що утворюються на поверхні ртутного електрода, будуть дифундувати в глиб ртуті, розчиняючись в ній з утворенням амальгами.
І на аноді та на катоді утворюються нові хімічні сполуки, яких раніше у розчині не було. Якщо відбувається перенесення заряду з однієї фази в іншу, то міжфазної межі встановлюється електричний потенціал (енергія).
Якщо електроди з'єднати провідником, то за достатньої різниці потенціалів між електродами опір розчину руху зарядів буде подолано і через розчин потече електричний струм (рух зарядів). Цей струм може бути виміряний.
Електрохімічні методи хімічного аналізу засновані на використанні явищ і процесів, що протікають на поверхні електрода, в приелектродному шарі або розчині електроліту, пов'язаних з хімічною природою і вмістом компонентів в розчині.
Вимірюють електричні властивості системи електрод – електроліт (потенціал електрода, силу електричного струму, кількість електрики, електричну провідність та ін.), всі розглянуті електричні величини залежать від концентрації будь-яких компонентів розчину електроліту. Отже, будь-яка з них - електрична провідність електроліту, потенціал електрода, сила електричного струму, ємність подвійного електричного шару та інші, може служити аналітичним сигналом, якщо вона функціонально пов'язана з концентрацією компонента, що визначається в аналізованому розчині і піддається вимірюванню. Виміряні значення електричних властивостей використовують для кількісного і іноді якісного хімічного аналізу складу речовини.
Існують різні класифікації електрохімічних методів для визначення концентрації компонента. Наприклад, методи можуть бути класифіковані в такий спосіб.
1. Методи, засновані на перебігу електродної реакції.
1.1. Методи, засновані на проходженні електричного струму через електрохімічний осередок:
-- вольтамперометрія– метод, заснований на вимірі сили дифузійного струму електроокислення або відновлення електровизначуваного компонента при певному значенні потенціалу індикаторного електрода;
- кулонометрія– метод, заснований на вимірі кількості електрики (закон Фарадея), витраченого на електрохімічну реакцію компонента, що визначається;
- електрогравіметрія– метод, заснований на вимірі маси компонента, виділеного на електроді при проходженні електричного струму через розчин електроліту (закон Фарадея);
1.2.Методи, засновані на вимірі різниці потенціалів між парою електродів при протіканні мізерно малих струмів у розчині:
-- потенціометрія– метод, заснований на вимірі різниці потенціалів індикаторного електрода та електрода порівняння;
2. Методи, не пов'язані з протіканням електродної реакції:
-- кондуктометрія– метод, заснований на вимірі питомої електричної провідності розчину, що залежить від природи та концентрації розчинених у ньому компонентів.
Концентрацію обумовленого компонента в пробі речовини об'єкта хімічного аналізу знаходять, як і в будь-якому іншому фізичному методі хімічного аналізу, з градуювального графіка.
Увага.Засоби вимірювання електричних властивостей речовин використовують також у хімічних методах кількісного хімічного аналізу, таких як титриметрія з метою фіксування при проведенні хімічної реакції еквівалентного обсягу титранта. Це так званий інструментальний (безіндикаторний) спосіб фіксування еквівалентності точки. За допомогою засобу вимірювання електричних властивостей речовин вимірюють відповідну електричну властивість компонента, що визначається, що змінюється при додаванні кожної порції титранта. У точці еквівалентності інтенсивність вимірюваної властивості різко змінюється і цей момент можна зафіксувати шляхом побудови та графічної обробки кривої титрування, побудованої в координатах “ виміряне значення електричної властивості – доданий обсяг титранта”. Концентрацію обумовленого компонента знаходять із закону еквівалентів. Це розширює можливості титриметричних методів в аналізі забарвлених, мутних розчинів, агресивних середовищ і т.д., де застосування кольорових індикаторів для фіксації точки еквівалентності неможливе. Методи титрування у разі називаються так: метод потенциометрического титрування, метод кондуктометрического титрування, метод амперометрического титрування тощо. За способом порівняння з еталоном ці методи належать до хімічних методів кількісного хімічного аналізу.
Характерними перевагами електрохімічних методів хімічного аналізу є низька межа визначення, експресність аналізу, легкість проведення вимірювань засобами вимірювання, можливість автоматизації та безперервності хімічного аналізу. Однак процеси, що відбуваються в електрохімічних осередках, досить складні для розуміння та інтерпретації отриманих результатів через їхню неоднозначність, тому цими методами практично неможливо провести якісний аналіз проби речовини, що обмежує можливості електрохімічних методів хімічного аналізу речовин.
Недоліком електрохімічних методів аналізу порівняно з хімічними методами кількісного аналізу є їхня порівняно невисока точність (похибка аналізу ~ 10%), однак деякі методи (кулонометрія, електрогравіметрія) відносяться до високоточних (похибка аналізу ~ 0,01%).
p align="justify"> Електрохімічні методи аналізу - це сукупність методів якісного і кількісного аналізу, заснованих на електрохімічних явищах, що відбуваються в досліджуваному середовищі або на межі розділу фаз і пов'язаних зі зміною структури, хімічного складу або концентрації аналізованої речовини.
Електрохімічні методи аналізу (ЕХМА) засновані на процесах, що протікають на електродах або міжелектродному просторі. Їх гідністю є висока точність і порівняльна простота як обладнання, так і методик аналізу. Висока точність визначається дуже точними закономірностями використовуваними в ЕХМА. Великою зручністю є те, що в цьому методі використовують електричні впливи, і те, що результат цього впливу (відгук) теж виходить у вигляді електричного сигналу. Це забезпечує високу швидкість і точність відліку, відкриває широкі можливості для автоматизації. ЕХМА відрізняються хорошою чутливістю і селективністю, в ряді випадків їх можна віднести до мікроаналізу, так як для аналізу іноді досить менше 1 мл розчину.
За різновидами аналітичного сигналу поділяють на:
1) кондуктометрію – вимірювання електропровідності досліджуваного розчину;
2) потенціометрію - вимірювання безструмового рівноважного потенціалу індикаторного електрода, для якого досліджувана речовина є потенціовизначальним;
3) кулонометрію – вимірювання кількості електрики, необхідної для повного перетворення (окислення або відновлення) досліджуваної речовини;
4) вольтамперометрію – вимірювання стаціонарних або нестаціонарних поляризаційних характеристик електродів у реакціях за участю досліджуваної речовини;
5) електрогравіметрію – вимірювання маси речовини, виділеної з розчину при електролізі.
27. Потенціометричний метод.
потенціометрію - вимірювання безструмового рівноважного потенціалу індикаторного електрода, для якого досліджувана речовина є потенціовизначальним.
А) стандартна (електрод порівняння) - має постійний потенціал, що не залежить від зовніш. умов
б) індивідуальний електрод - його потенціал залежить від концентрації речовини.
Потенціал залежить від концентрації: Е = f(c)
Рівняння Нериста Е = Е ° + lna kat
E° - Стандарт. Електрон. Потенціал (const)
R- Універ. Газова постійнаconst)
Т - абсолютна темп (t)- +273 °
.п - Число електронів участвующ. В окис./вост. Реакції
. а – активна концентрація
Метод потенціометрії
Іонометрія потенціометрування (до дослід. Р-ру невелик. Порціями додається стандарт.р-р(титран), після кожного додатку вимірюють потенціал.- Е)
Точка еквівалентності
ЕСх Vх = lт * Vт
28. Кондуктометричний метод.
кондуктометрія-вимірювання електропровідності досліджуваного розчину.
Кондуктометричне титрування
Кондуктометр (прилад)
Кондуктометричний аналіз (кондуктометрія) заснований на використанні залежності між електропровідністю (електричною провідністю) розчинів електролітів та їх концентрацією.
Про електропровідність розчинів електролітів - провідників другого роду - судять на підставі вимірювання їх електричного опору в електрохімічному осередку, який є скляною посудиною (склянкою) з двома впаяними в неї електродами, між якими і знаходиться випробуваний розчин електроліту. Через комірку пропускають змінний електричний струм. Електроди найчастіше виготовляють з металевої платини, яку збільшення поверхні електродів покривають шаром губчастої платини шляхом електрохімічного осадження з розчинів платинових сполук (електроди з платинованої платини).
29.Полярографія.
Полярографія - метод якісного і кількісного хімічного аналізу, заснований на отриманні кривих залежності величини струму від напруги в ланцюзі складається з досліджуваного розчину і занурених в нього електродів, один з яких сильно поляризується, а інший практично неполяризується. Отримання таких кривих – полярограм – виробляють за допомогою полярографів.
Полярографічний метод характеризується великою чутливістю. Для виконання аналізу зазвичай достатньо 3-5 мл досліджуваного розчину. Аналіз за допомогою автореєструючого полярографа триває лише близько 10 хвилин. Полярографію використовують для визначення в об'єктах біологічного походження вмісту отруйних речовин (наприклад, сполук ртуті, свинцю, талію та ін.), для визначення ступеня насичення крові киснем, дослідження складу повітря, що видихається, шкідливих речовин у повітрі промислових підприємств. Полярографічний метод аналізу має велику чутливість і дає можливість визначати речовини за дуже незначної (до 0,0001%) концентрації їх у розчині.
30.Класифікація спектральних методів аналізу. Концепція спектру.
Спектральний аналіз - це сукупність методів визначення кач.і кількіс. Складу, а також структури речовини (заснованих на взаємодії дослід.об'єкта з різними типами випромінювання.)
Усі спектроскопічні методи засновані на взаємодії атомів, молекул або іонів, що входять до складу речовини, що аналізується, з електромагнітним випромінюванням. Ця взаємодія проявляється у поглинанні чи випромінюванні фотонів (квантів). Залежно від характеру взаємодії проби з електромагнітним випромінюванням виділяють дві групи методів
Емісійні та абсорбційні. Залежно від того, які частинки формують аналітичний сигнал, розрізняють методи атомної спектроскопії та методи молекулярної спектроскопії.
Емісійна
В емісійних методах аналізована проба внаслідок її збудження випромінює фотони.
абсорбційна
В абсорбційних методах випромінювання стороннього джерела пропускають через пробу, при цьому частина квантів вибірково поглинається атомами чи молекулами
Спектр- Розподіл значень фізичної величини (зазвичай енергії, частоти або маси). Графічне уявлення такого розподілу називається спектральною діаграмою. Зазвичай під спектром мається на увазі електромагнітний спектр - спектр частот (або те саме, що енергій квантів) електромагнітного випромінювання.
1.відображення світла
2. поворот пучка світла (дефракція)
3.розсіювання світла: нефелометрія,турбідиметрія
4.поглинання світла
5перевипромінювання
А) фосфоресценція (триває довго)
Б) флуоресценція (дуже коротка)
За характером розподілу значень фізичної величини спектри можуть бути дискретними (лінійчастими), безперервними (суцільними), а також представляти комбінацію (накладення) дискретних та безперервних спектрів.
Прикладами лінійних спектрів можуть служити мас-спектри та спектри зв'язано-пов'язаних електронних переходів атома; прикладами безперервних спектрів - спектр електромагнітного випромінювання нагрітого твердого тіла та спектр вільно-вільних електронних переходів атома; прикладами комбінованих спектрів - спектри випромінювання зірок, де суцільний спектр фотосфери накладаються хромосферні лінії поглинання чи більшість звукових спектрів.
31.Фотометрія: принцип методу, застосування в суд.
Фотометрія – спектральний метод заснований на поглинанні електромагнітного випромінювання видимого та ближнього ультрафіолетового діапазону (метод заснований на поглинанні світла)
Молекулярна атомна
Спектроскопія Спектроскопія (В електрон.Аналізі)
Кювета – через неї пропускають світло
l
I (інтенсивність вихід. світла)
I° – інтенсивність падаючого світла.
Фотометрія – розділ фізичної оптики та вимірювальної техніки, присвячений методам дослідження енергетичних характеристик оптичного випромінювання у процесі його випромінювання, поширення у різних середовищах та взаємодії з тілами. Фотометрію проводять у діапазонах інфрачервоного (довжини хвиль – 10 –3…7 10 –7 м), видимого (7 10 –7…4 10 –7 м) та ультрафіолетового (4 10 –7…10 –8 м) оптичних випромінювань. При поширенні електромагнітного випромінювання оптичного діапазону в біологічному середовищі спостерігаються ряд основних ефектів: поглинання та розсіювання випромінювання атомами та молекулами середовища, розсіювання на частинках неоднорідностей середовища, деполяризація випромінювання. Реєструючи дані взаємодії оптичного випромінювання із середовищем, можна визначити кількісні параметри, пов'язані з медико-біологічними характеристиками об'єкта, що досліджується. Для виміру фотометричних величин застосовують прилади – фотометри. З погляду фотометрії, світло - це випромінювання, здатне викликати відчуття яскравості при впливі на людське око. В основі фотометрії як науки лежить розроблена А. Гершуном теорія світлового поля.
Існують два загальні методи фотометрії: 1) візуальна фотометрія, в якій при вирівнюванні механічними або оптичними засобами яскравості двох полів порівняння використовується здатність ока людини відчувати відмінності в яскравості; 2) фізична фотометрія, у якій порівняння двох джерел світла використовуються різні приймачі світла іншого роду – вакуумні фотоелементи, напівпровідникові фотодіоди тощо.
32. Закон Бугера-Ламберта-Бера, його використання в кількісному аналізі.
Фізичний закон, що визначає ослаблення паралельного монохроматичного пучка світла при поширенні його в поглинаючому середовищі.
Закон виражається такою формулою:
![]() ,
,

де інтенсивність вхідного пучка, товщина шару речовини, через яке проходить світло, показник поглинання (не плутати з безрозмірним показником поглинання, який пов'язаний сформулою, де довжина хвилі).
Показник поглинання характеризує властивості речовини і залежить від довжини хвилі λ світла, що поглинається. Ця залежність називається спектром поглинання речовини.
Для розчинів поглинаючих речовин в розчинниках, що не поглинають світло, показник поглинання може бути записаний як
де - коефіцієнт, що характеризує взаємомолекули поглинаючої розчиненої речовини зі світлом з довжиною хвилі λ, -концентрація розчиненої речовини, моль/л.
Твердження, що не залежить від, називається законом Бера (не плутати з законом Бера). Цей закон передбачає, що на здатність молекули поглинати світло не впливають інші навколишні молекули цієї ж речовини в розчині. Однак, спостерігаються численні відхилення від цього закону, особливо при великих.
Якщо через деякий шар розчину або газу товщиною (проходить світловий потік інтенсивністю I, то за законом Ламберта - Бера кількість поглиненого світла буде пропорційно інтенсивності /, концентрації з речовини, що поглинає світло, та товщині ШАРУ) закон БМБ, який пов'язує інтенсивності світла, що падає на Ну це так само, як заломлення, тільки згасання в речовині. Яке світло поглинає під певним відсотком. Тобто залишок від виходу світла є
33.ІЧ-спектроскопія.
Цей метод аналізу ґрунтується на записі інфрачервоних спектрів поглинання речовини. Поглинання речовиною в області інфрачервоного випромінювання відбуваються за рахунок коливань атомів в молекулах. Коливання поділяються на валентні (коли під час коливання змінюються відстані між атомами) і коливальні (коли під час коливання змінюються кути між зв'язками). Переходи між різними коливальними станами в молекулах квантовані, завдяки чому поглинання ІЧ-області має форму спектра, де кожному коливанню відповідає своя довжина хвилі. Зрозуміло, що довжина хвилі для кожного коливання залежить від того, які атоми в ньому беруть участь, і крім того, вона мало залежить від їх оточення.
метод ІЧ-спектроскопії не є розділяючим методом, тобто при дослідженні будь-якої речовини може виявитися, що насправді досліджувалася суміш декількох речовин, що звичайно сильно спотворить результати розшифровки спектру. Ну і все ж говорити про однозначну ідентифікацію речовини за допомогою методу ІЧ-спектроскопії не цілком правильно, оскільки метод швидше дозволяє виявити певні функціональні групи, а не їх кількість у поєднанні та їх спосіб зв'язку один з одним.
Метод ІЧ-спектроскопії використовується при проведенні досліджень полімерних матеріалів, волокон, лакофарбових покриттів, наркотичних засобів (при ідентифікації наповнювача як часто виступають вуглеводи в тому числі полісахариди). Особливо метод незамінний для дослідження мастильних матеріалів, тим що дає можливість одночасного визначення природи як основи мастильного матеріалу, і можливих добавок (присадок) до цієї основі.
34. Рентгенофлуоресцентний аналіз.
(РФА) - один із сучасних спектроскопічних методів дослідження речовини з метою отримання його елементного складу, тобто його елементного аналізу. За допомогою нього можуть аналізуватись різні елементи від берилію (Be) до урану (U). Метод РФА заснований на зборі та подальшому аналізі спектра, отриманого шляхом впливу на досліджуваний матеріал рентгенівським випромінюванням. При опроміненні атом перетворюється на збуджений стан, що полягає у переході електронів більш високі енергетичні рівні. У збудженому стані атом перебуває вкрай малий час, близько однієї мікросекунди, після чого повертається у спокійне становище (основний стан). При цьому електрони із зовнішніх оболонок або заповнюють вакантні місця, що утворилися, а надлишок енергії випромінюється у вигляді фотона, або енергія передається іншому електрону із зовнішніх оболонок (оже-електрон)
Екологія та охорона навколишнього середовища: визначення важких металів у ґрунтах, опадах, воді, аерозолях та ін.
Геологія та мінералогія: якісний та кількісний аналіз ґрунтів, мінералів, гірських порід та ін.
Металургія та хімічна індустрія: контроль якості сировини, виробничого процесу та готової продукції
Лакофарбова промисловість: аналіз свинцевих фарб
35. Атомно-емісійна спектроскопія.
Атомно-емісійний спектральний аналіз - це сукупність методів елементного аналізу, заснованих на вивченні спектрів випромінювання вільних атомів та іонів у газовій фазі. Зазвичай емісійні спектри реєструють найбільш зручної оптичної області довжин хвиль від 200 до 1000 нм.
АЕС (атомно-емісійна спектрометрія) – спосіб визначення елементного складу речовини за оптичними спектрами випромінювання атомів та іонів аналізованої проби, що збуджується в джерелах світла. Як джерела світла для атомно-емісійного аналізу використовують полум'я пальника або різні види плазми, включаючи плазму електричної іскри або дуги, плазму лазерної іскри, індуктивно-пов'язану плазму, тліючий розряд та ін. АЕС – найпоширеніший експресний високочутливий метод ідентифікації та кількісного визначення домішок у газоподібних, рідких та твердих речовинах, у тому числі й у високочистих.
Області застосування:
Металургія: аналіз складу металів та сплавів,
Гірничодобувна промисловість: дослідження геологічних зразків та мінеральної сировини,
Екологія: аналіз води та ґрунту,
Техніка: аналіз моторних масел та ін. технічних рідин на домішки металів,
Біологічні та медичні дослідження.
Принцип дії.
Принцип дії атомно-емісійного спектрометра є досить простим. Він заснований на тому, що атоми кожного елемента можуть випромінювати світло певних довжин хвиль - спектральні лінії, причому ці довжини хвиль різні для різних елементів. Для того щоб атоми почали випромінювати світло, їх необхідно порушити - нагріванням, електричним розрядом, лазером або іншим способом. Чим більше атомів даного елемента є в аналізованому зразку, тим яскравіше буде випромінювання відповідної довжини хвилі.
Інтенсивність спектральної лінії аналізованого елемента, крім концентрації аналізованого елемента, залежить від великої кількості різних факторів. З цієї причини розрахувати теоретично зв'язок між інтенсивністю лінії та концентрацією відповідного елемента неможливо. Ось чому для проведення аналізу необхідні стандартні зразки, близькі за складом до проби, що аналізується. Попередньо ці стандартні зразки експонуються (пропалюються) на приладі. За результатами цих пропалів кожному за аналізованого елемента будується градуювальний графік, тобто. залежність інтенсивності спектральної лінії елемента від концентрації. Згодом, при проведенні аналізу проб, за цими градуювальними графіками і проводиться перерахунок вимірюваних інтенсивностей в концентрації.
Підготовка проб аналізу.
Слід мати на увазі, що реально аналізу піддається кілька міліграмів проби з її поверхні. Тому для отримання правильних результатів проба має бути однорідною за складом і структурою, при цьому склад проби повинен бути ідентичним складу аналізованого металу. При аналізі металу у ливарному чи плавильному виробництві для виливки проб рекомендується використовувати спеціальні кокілі. При цьому форма проби може бути довільною. Необхідно лише, щоб аналізований зразок мав достатню поверхню і міг бути затиснутий у штативі. Для аналізу дрібних зразків, наприклад прутків чи дроту, можуть бути використані спеціальні адаптери.
Переваги методу:
Безконтактність,
Можливість одночасного кількісного визначення великої кількості елементів,
Висока точність,
Низькі межі виявлення,
Простота пробопідготовки,
Низька собівартість.
36. Атомно-абсорбційна спектроскопія.
метод количества. визначення елементного складу досліджуваної речовини по атомним спектрам поглинання, заснований на здатності атомів вибірково поглинати електромагнітне випромінювання в разл. дільницях спектра. A.-a.a. проводять на спец. приладах – абсорбц. спектрофотометрів. Пробу аналізованого матеріалу розчиняють (зазвичай з утворенням солей); розчин у вигляді аерозолю подають у полум'я пальника. Під дією полум'я (3000 ° C) молекули солей дисоціюють на атоми, які можуть поглинати світло. Потім через полум'я пальники пропускають пучок світла, у спектрі якого є відповідні тому чи іншому елементу спектральні лінії. Із загального випромінювання досліджувані спектральні лінії виділяють монохроматором, а їх інтенсивність фіксують блоком реєстрації. Матем. обробка проводиться за формулою: J = J0 * e-kvI,
де J і J0, - Інтенсивності минулого і падаючого світла; kv - Коеф. поглинання, що залежить від його частоти; I - товщина поглинаючого шару
більш чутливий, ніж АЕС
37. Нефелометрія та турбідиметрія.
S = lg (I°/I) інтенсивність падаюч. У р-р(I°) ділимо на інтенсивність вихід з р-ра(I) =
k-const каламутності
b – довжина шляху пучка світла
N-число частинок од. р-ра
У нефелометричному та турбідиметричному аналізі використовується явище розсіювання світла твердими частинками, що знаходяться у розчині у зваженому стані.
Нефелометрія - метод визначення дисперсності та концентрації колоїдних систем за інтенсивністю розсіяного ними світла. Нефелометрія, вимірювання виробляються у спеціальному приладі нефелометрі, дія якого заснована на порівнянні інтенсивності розсіяного досліджуваним середовищем світла з інтенсивністю світла, розсіяного іншим середовищем, що є стандартом. Теорія розсіювання світла колоїдними системами, в яких розміри частинок не перевищують довжини напівхвилі падаючого світла, була розроблена англійським фізиком Дж. Релеєм в 1871. За законом Релея, інтенсивність світла I, розсіяного в напрямку, перпендикулярному до променя, що падає, де q - інтенсивність падаючого світла, N - загальна кількість частинок в одиниці об'єму, або часткова концентрація, v - обсяг однієї частинки, \ - довжина хвилі падаючого світла, k - константа, яка залежить від показників заломлення колоїдних частинок і навколишнього дисперсійного середовища, відстані від джерела світла, а також від прийнятих одиниць виміру
Турбідиметрія – метод аналізу каламутних середовищ, заснований на вимірі інтенсивності поглиненого ними світла. Турбідиметричні вимірювання проводять у світлі за допомогою турбідиметрів візуальних або фотоелектричних колориметрів. Методика вимірювань аналогічна колориметричній і ґрунтується на застосовності до каламутних середовищ Бугера-Ламберта - закону Бера, який у разі суспензій справедливий лише для дуже тонких шарів або при значних розбавленнях. При турбідиметрії потрібно ретельне дотримання умов утворення дисперсної фази, аналогічних умов, що дотримуються при нефелометрії. Значне удосконалення турбідиметрії полягає у застосуванні турбідиметричного титрування по максимуму помутніння за допомогою фотоелектричних колориметрів. Турбідиметрія з успіхом використовуються для аналітичного визначення сульфатів, фосфатів, хлоридів, ціанідів, свинцю, цинку та ін.
Основною перевагою нефелометричних і турбідиметричних методів є їхня висока чутливість, що особливо цінно по відношенню до елементів або іонів, для яких відсутні кольорові реакції. У практиці широко застосовується, наприклад, нефелометричне визначення хлориду та сульфату у природних водах та аналогічних об'єктах. За точністю турбідиметрія і нефелометрія поступаються фотометричним методам, що пов'язано, головним чином, з труднощами отримання суспензій, що володіють однаковими розмірами частинок, стабільністю в часі і т. д. До звичайних порівняно невеликих похибок фотометричного визначення додаються помилки, пов'язані з недостатньою відтворюваністю. властивостей суспензій
Нефелометрію і турбідиметрію застосовують, напр., для визначення SO4 у вигляді суспензії BaSO4, Сl-у вигляді суспензіи AgCl, S2-у вигляді суспензії CuS з ниж. межами визначених вмістів ~ 0,1 мкг/мл. Для стандартизації умов аналізу експериментах необхідно суворо контролювати т-ру, обсяг суспензії, концентрації реагентів, швидкість перемішування, час проведення вимірів. Осадження повинне протікати швидко, а частинки, що осаджуються, повинні мати малі розміри і низьку р-римість. Для запобігання коагуляції великих частинок в розчин часто додають стабілізатор, напр. желатин, гліцерин.
38. Хроматографія: історія виникнення, принцип методу, застосування суду. Дослідження.
Хроматографія - динамічний сорбційний метод поділу та аналізу сумішей речовин, а також вивчення фізико-хімічних властивостей речовин. Заснований на розподілі речовин між двома фазами - нерухомою (тверда фаза або рідина, пов'язана на інертному носії) та рухомою (газова або рідка фаза, елюенти). Назва методу пов'язана з першими експериментами з хроматографії, у ході яких розробник методу Михайло Колір розділяв яскраво забарвлені рослинні пігменти.
Метод хроматографії був вперше застосований російським вченим-ботаніком Михайлом Семеновичем Цвєтом у 1900 році. Він використовував колонку, заповнену карбонатом кальцію, для поділу пігментів рослинного походження. Перше повідомлення про розробку методу хроматографії було зроблено Кольором 30 грудня 1901 на XI З'їзді дослідників природи та лікаріву С.-Петербурзі. Першу друковану роботу з хроматографії було опубліковано в 1903 році, в журналі Праці Варшавського товариства дослідників природи. Вперше термін хроматографіяз'явився у двох друкованих працях Кольори у 1906 році, опублікованих у німецькому журналі Berichte der Deutschen Botanischen Gesellschaft. У 1907 році Колір демонструє свій метод Німецькому Ботанічному товариству.
У 1910-1930 роки метод був незаслужено забутий і мало розвивався.
У 1931 році Р. Кун, А. Вінтерштейн та Е. Ледерер за допомогою хроматографії виділили з сирого каротину α та β фракції у кристалічному вигляді, чим продемонстрували препаративну цінність методу.
У 1941 році А. Дж. П. Мартін і Р. Л. М. Сінг розробили новий різновид хроматографії, в основу якої лягла відмінність в коефіцієнтах розподілу речовин, що розділяються між двома рідинами, що не змішуються. Метод отримав назву « розподільна хроматографія».
У 1947 році Т. Б. Гапон, Є. Н. Гапон та Ф. М. Шемякін розробили метод «іонообмінної хроматографії».
У 1952 році Дж. Мартіну та Р. Сінгу було присуджено Нобелівську премію в галузі хімії за створення методу розподільчої хроматографії.
З середини XX століття і до наших днів хроматографія інтенсивно розвивалася і стала одним із найбільш широко застосовуваних аналітичних методів.
Класифікація: Газова, Рідина
Основи хроматографіч. процесу.Для проведення хроматографіч. поділу в-в або визначення їх фіз.-хім. Показників зазвичай використовують спец. прилади – хроматографи. основ. вузли хроматографа - хроматографіч. стовпчик, детектор, а також пристрій для введення проби. Колонка, що містить сорбент, виконує ф-цію поділу аналізованої суміші на складові компоненти, а детектор -ф-цію їх кількостей. визначення. Детектор, розташований на виході з колонки, автоматично безперервно визначає концентрацію з'єднань, що розділяються. в потоці рухомий Після введення аналізованої суміші з потоком рухомої фази в колонку зони всіх в-в розташовані на початку хроматографіч. колонки (рис. 1). Під впливом потоку рухомий фази компоненти суміші починають переміщатися вздовж колонки з разл. швидкостями, величини яких брало назад пропорційні коефіцієнтам розподілу До хроматографованих компонентів. Добре сорбируемые в-ва, значення константрозподілу для яких брало великі, пересуваються вздовж шару сорбенту по колонці повільніше, ніж погано сорбируемые. Тому найшвидше з колонки виходить компонент А, потім компонент Б і останнім залишає колонку компонент В (К А<К Б <К В). Сигнал детектора, величина к-рого пропорциональна концентрации определяемого в-ва в потоке элюента, автоматически непрерывно записывается и регистрируется (напр., на диаграммной ленте). Полученная хроматограмма отражает расположение хроматографич. зон на слое сорбента или в потоке подвижной фазы во времени.

Мал. 1.Поділ суміші з трьох компонентів (А, Б і В) на хроматографічній колонці К з детектором Д: а - положення хроматографічних зон компонентів, що розділяються в колонці через певні інтервали часу; б – хроматограма (С – сигнал, t – час) .
При плоскошаровому хроматографіч. розділення аркуш паперу або пластину з шаром сорбенту з нанесеними пробами досліджуваного в-ва поміщають в хроматографіч. камери. Після поділу компоненти визначають будь-яким відповідним методом.
39. Класифікація хроматографічних методів.
Храмотографія - метод поділу та аналізу речовин, заснований на розподілі аналіз. В-ва між 2 фазами: рухомий та нерухомий
Розчин суміші речовин, що підлягають поділу, пропускають через скляну трубку(Адсорбційну колонку) заповнену адсорбентом. В результаті компоненти суміші утримуються на різній висоті стовпа адсорбенту у вигляді окремих зон (шарів). Реч-ва краще адсорбір. Нах вгору частини стовпа, а гірше адсорбуються в нижній частині стовпа. В-ва не здатні адсорбуватися - проходять через колонку не затримуючись і збираються у фільтрі.
Класифікації:
1. За агрегатним станом фаз.
1) Рухлива
А) газова (інертні гази: гелій, аргон, азон)
Б)рідинна
2. за способом проведення
1) на площині (планарна); паперова тонкошарова
2) колонкова
А) насадочна(насадкова колонка наповнена сорбентом)
Б) капілярна (тонкий скляний/кварцовий капіляр на внутр.поверхні якого нанесена нерухома фаза)
Можна опр. Реч-ва в небольш.кол-вах.
Летючі в-ва поділяються.
40. Хроматограма. Основні параметри хроматограф.піка.
Хроматограма - результат реєстрації залежності концентрації компонентів на виході з колонки від часу.
H S
Кожен пік на хроматограмі характеризується двома основними параметрами
1. Час утримування ( t R) - це час від моменту введення аналізованої проби до моменту реєстрації максимуму хроматографічного піку. Воно залежить від природи речовини та є якісною характеристикою.
2. Висота ( h) або площа ( S) піка
S = ½ ω × h. (4)
Висота та площа піку залежать від кількості речовини та є кількісними характеристиками.
Час утримування складається з двох складових - часу перебування речовин у рухомій фазі ( t m) та часу перебування в нерухомій фазі ( t s):
Ідентифікацію піків невідомих компонентів аналізованої суміші проводять шляхом зіставлення (порівняння) відносить. величин, що визначаються безпосередньо з хроматограми, з відповідними табличними даними для відомих сполук. При ідентифікації хроматографії достовірний тільки заперечують. відповідь; напр., пік i не є в-вом А, якщо часи утримування піку i і в-ва А не збігаються. Збіг часів утримування піку i і в-ва А - необхідна, але недостатня умова для висновку, що пік i - це в-во А.
У практичній роботі вибір того чи іншого параметра для кількісної розшифровки хроматограм визначається сукупним впливом декількох факторів швидкістю та зручністю розрахунку, формою (широкий, вузький) та ступенем асиметрії хроматографічного піку, ефективністю використовуваної колонки, повнотою поділу компонентів суміші, наявністю необхідних автоматизованих пристроїв. комп'ютерних систем обробки даних хроматографічного аіалізу).
Визначається параметр хроматографічного піку вимірюється оператором на хроматограмі вручну після закінчення циклу поділу компонентів аналізованої суміші
Визначається параметр хроматографічного піку вимірюється автоматично за допомогою цифрових вольтметрів, інтеграторів або спеціалізованих ЕОМ одночасно з поділом компонентів аналізованої суміші в колонці та записом хроматограми
Оскільки техніка розшифровки хроматограм зводиться до вимірювання параметрів хроматографічних піків цікавого і стандартного з'єднань, умови хроматографування повинні забезпечувати по можливості повне їх поділ всі інші складові вихідної проби в умовах аналізу можуть не відокремлюватися один від одного або навіть взагалі не виявлятися на хроматограмі (у цьому полягає перевага методу внутрішнього стандарту перед методом внутрішньої нормалізації)
41. Якісний хроматографіч.аналіз.
При достатній довжині колонки можна зробити повний поділ компонент будь-якої суміші. А після елюювання розділених компонентів окремі фракції (елюати) визначити кількість компонентів суміші (воно відповідає кількості елюатів), встановити їх якісний склад, визначити кількість кожного з них, використавши відповідні методи кількісного аналізу.
Якісний хроматографічний аналіз, тобто. індетифікація речовини за його хроматограмою, може бути виконаний порівнянням хроматограїчних характеристик, найчастіше утримуваного об'єму (тобто об'єму рухомої фази, пропущеної через колонку від початку введення суміші до появи даного компонента на виході з колонки), знайдених за певних умов для аналізованої компоненти суміші та для еталона.
42.Кількісний хроматограф.аналіз.
Кількісний хроматографічний аналіз зазвичай проводять на хроматографі. Метод заснований на вимірі різних параметрів хроматографічного піку, що залежать від концентрації хроматографованих речовин - висоти, ширини, площі і об'єму, що утримується, або добутку утримуваного об'єму на висоту піку.
У кількісній газовій хроматографії застосовують методи абсолютної градуювання та внутрішньої нормалізації, або нормування. Використовується метод внутрішнього стандарту. При абсолютному градуюванні експериментально визначають залежність висоти або площі піку від концентрації речовини і будують графіки градуювання або розраховують відповідні коефіцієнти. Далі визначають ті ж характеристики піків в аналізованій суміші, і за градуювальним графіком знаходять концентрацію аналізованої речовини. Цей простий і точний метод є основним щодо мікродомішок.
При використанні методу внутрішньої нормалізації приймають суму будь-яких параметрів піків, наприклад, суму висот усіх піків або суму їх площ за 100%. Тоді відношення висоти окремого піку до суми висот або відношення площі одного піку до суми площ при множенні на 100 характеризуватиме масову частку (%) компонента в суміші. При такому підході необхідно, щоб залежність величини параметра, що вимірюється від концентрації була однаковою для всіх компонентів суміші.
43. Планарна хроматографія. Використання тонкошарової хроматографії для аналізу чорнила.
Першою формою використання целюлози в тонкошаровій хроматографії була паперова хроматографія. Доступні пластинки для ТСХ і високопродуктивної ТСХ дозволяють розділяти суміші полярних речовин, при цьому як елюент використовуються, принаймні, потрійні суміші з води, органічного розчинника, що не змішується з нею, і водорозчинного розчинника, що сприяє утворенню однієї фазиү окисл // [ віднов] ү восст)),
R - Універсальна газова постійна, рівна 8,31 Дж / (моль. К); Т – абсолютна температура; F - Постійна Фарадея (96500 Кл/моль); n - Число електронів, що беруть участь в електродній реакції; а окис, а віднов- активності відповідно окисленої та відновленої форм редокс-системи;[ окисл] і[ віднов] - їх молярні концентрації; - окис, - відновлення - коефіцієнти активності; Е° – стандартний потенціал редокс-системи.
Підставляючи Т= 298,15 К та числові значення констант у рівняння, отримуємо:
Е = Е°+ (0,059/ n) lg (а окис/а віднов)
Е = Е°+ (0,059/ n) lg ([ окисл] ү окисл / ([ віднов] ү восст))
Методи прямої потенціометріїзасновані на застосуванні рівняння Нернста для знаходження активності або концентрації учасника електродної реакції експериментально виміряної ЕРС ланцюга або потенціалу електрода. Найбільшого поширення серед прямих потенціометричних методів набув метод визначення рН, але створення останнім часом надійно працюючих іоноселективних електродів значно розширило практичні можливості прямих методів. Показник рН вимірюють методом і потенціометричного титрування.
Для визначення рН найчастіше використовують скляний електрод. Основними перевагами скляного електрода є простота роботи, швидке встановлення рівноваги та можливість визначення рН в окислювально-відновних системах. До недоліків відносяться крихкість матеріалу електрода та складність роботи при переході до сильнолужних та сильнокислих розчинів.
Крім концентрації іонів водню, прямим потенціометричним методом з іоноселективними електродами можна визначити вміст кількох десятків різних іонів.
Потенціометричне титруванняґрунтується на визначенні точки еквівалентності за результатами потенціометричних вимірів. Поблизу точки еквівалентності відбувається різка зміна потенціалу індикаторного електрода. Так само, як і в інших титриметричнихметоди, реакції потенціометричного титрування повинні протікати суворо стехіометричномати високу швидкість і йти до кінця.
Для потенціометричного титрування збирають ланцюг з індикаторного електрода в аналізованому розчині та порівняння електрода. Як електроди порівняння найчастіше використовують каломельний або хлорсрібний електроди.
Тип індикаторного електрода при потенціометричному титруванні залежить від властивостей титриметричноїсуміші та її взаємодії з електродом. У кислотно-основному титруванні використовують скляний електрод, в окислювально-відновному - інертний (платиновий) електрод або електрод, оборотний по відношенню до одного з іонів, що містяться в титриметриметричноїсуміші; в осаді - срібний електрод; в комплексонометричному- металевий електрод, оборотний до іону металу, що титрується.
Для знаходження точки еквівалентності часто будують диференціальну криву в координатах DЕ/ D V - V . На точку еквівалентності вказує максимум отриманої кривої, а відлік по осі абсцис, що відповідає цьому максимуму, дає об'єм титранта витраченоготитрування до точки еквівалентності. Визначення точки еквівалентності до диференціальної кривої значно точніше, ніж за простою залежністю Е - V.
Основними перевагами методу потенціометричного титрування є висока точність і можливість проводити визначення розбавлених розчинах, в каламутних і пофарбованих середовищах, а також визначати кілька речовин в одному розчині без попереднього поділу. Значно розширюється сфера практичного застосування потенціометричного титрування при використанні неводних розчинників. Вони дозволяють аналізувати багатокомпонентні системи, які у водному розчині визначити не вдається, провести аналіз речовин, які не розчиняються або розкладаються у воді, і т. д. Потенціометричне титрування легко може бути автоматизовано. Промисловість випускає кілька типів автотитраторів, що використовують потенціометричні датчики.
До недоліків потенціометричного титрування можна віднести не завжди швидке встановлення потенціалу після додавання титранта та необхідність у багатьох випадках проводити при титруванні велику кількість відліків.
У потенціометричному аналізі основними вимірювальними приладами є різні потенціометри типів. Вони призначені для вимірювання ЕРС електродної системи. Так як ЕРС залежить від активності відповідних іонів у розчині, багато потенціометрів дозволяють безпосередньо вимірювати також величину рХ - негативний логарифм активності іона Х. Такі потенціометри в комплекті з відповідним іоноселективним електродом носять назву іономірів. Якщо потенціометр та електродна система призначені для вимірювання активності лише водневих іонів, прилад називається рН-метром.А.А. Віксарєв, С.А. Зуйкова, Н.А. Чемеріс, Н.Г. Доміна